内毒素控制、PUPSIT滤器完整性测试要求、一次性系统颗粒物控制、注射用水超低TOC控制带来的挑战与应对策略
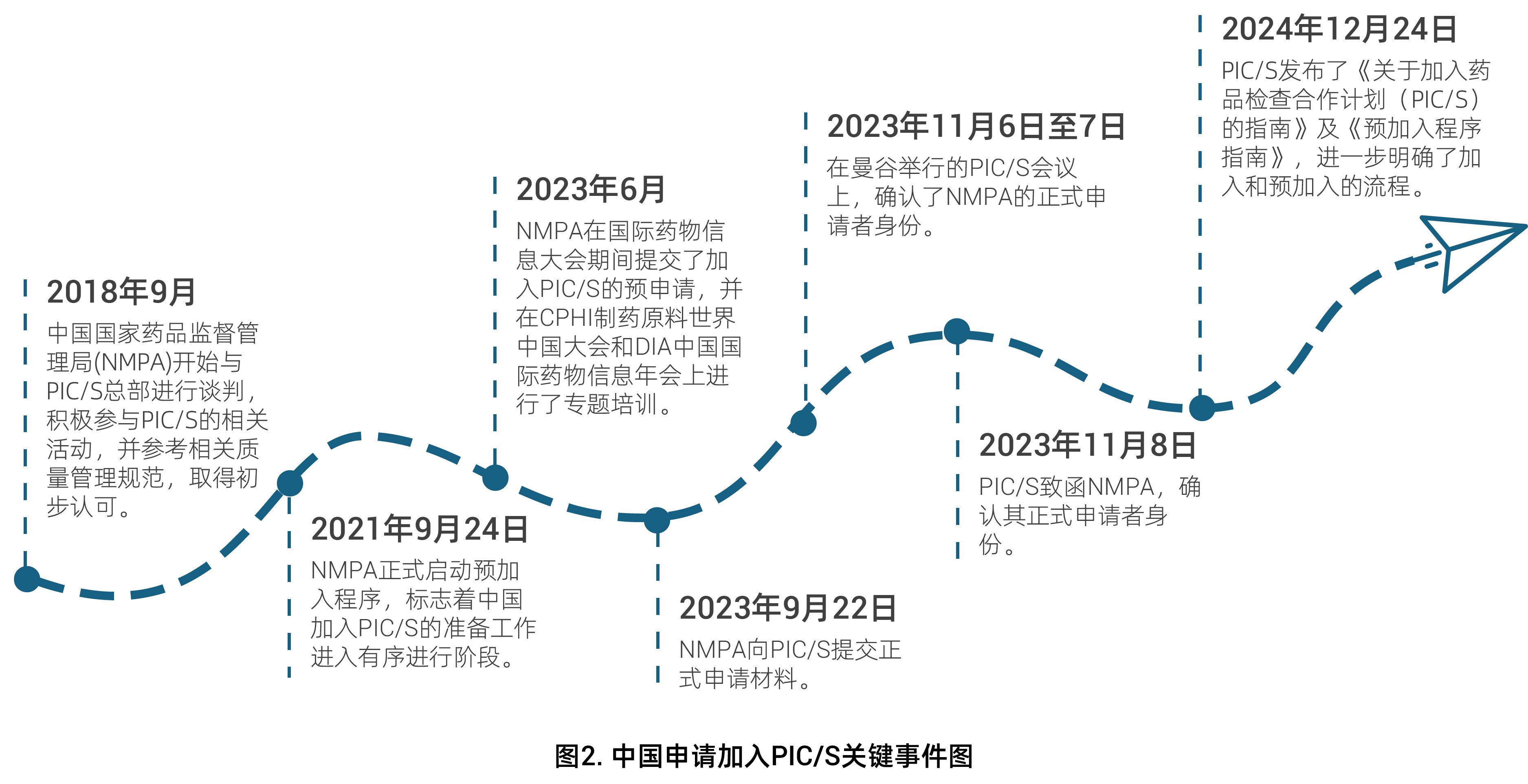
2023年9月,国家药品监督管理局正式向国际药品检查合作计划(PIC/S)提交加入申请,并于同年11月8日成功获得正式申请者资格。2025年3月17日,药监局发布《药品生产质量管理规范(2010年修订)》无菌药品附录征求意见稿,该技术文件与欧盟GMP无菌附录Annex 1保持高度协调一致。这一系列举措标志着我国药品监管体系与国际标准接轨取得实质性进展,将显著提升中国药品监管的国际认可度,为国产药品开拓国际市场创造有利条件。
然而,中国药企在把握国际化机遇的同时,也面临着更为严苛的技术挑战。特别是在污染控制策略(CCS)方面,国际标准对以下关键环节提出了更高要求:内毒素控制需建立更严格的标准和监测体系;除菌过滤器使用前必须实施灭菌后完整性测试(PUPSIT)以确保无菌保证;微粒污染控制需要建立更精细的工艺控制方案;注射用水储存过程中的总有机碳(TOC)控制也需达到更严苛的标准。这些技术要求的提升,既是对企业质量管理体系的考验,也是推动产业升级的重要契机。
挑战一:内毒素控制再升级- AEX膜层析技术攻克难关
随着PIC/S对注射剂/生物制品细菌内毒素管控标准的持续提升,传统生产工艺已难以满足痕量内毒素残留的严苛要求。
内毒素(Endotoxin)作为革兰氏阴性菌细胞壁外膜的主要成分,是一种具有强致热活性的脂多糖(LPS),在细菌死亡或裂解后释放至环境中。从分子特性来看,内毒素单体含有带负电荷的磷酸基和羧基,其等电点约为2。这一特性使得在pH>2的溶液中,内毒素始终维持电负性状态,这也为基于电荷作用的离子交换层析法(IEX)提供了去除热原的理论基础。相较于传统树脂基阴离子交换层析(AEX)技术,科百特创新研发的Purcise™ Q强离子交换吸附膜(简称Q膜)展现出显著的技术优势:具有高载量、高流速特性,支持从实验室研发到工业化生产的全流程扩展需求;即用型设计大幅简化操作流程,同时保持高效的内毒素去除效能,为生物制药企业提供了更高效、更可靠的除热原解决方案。

在实际生产过程中,当采用10kD膜包进行蛋白浓缩时,样品及洗滤Buffer中的内毒素会随超滤过程不断富集。更值得注意的是,浓缩过程中内毒素与蛋白的吸附作用会显著增强,导致最终产物中的内毒素难以有效去除。科百特Purcise™ Q膜吸附器通过创新的离子交换技术,可实现对Buffer中高浓度内毒素的有效控制(见图5 Case 1),显著降低浓缩工艺中的内毒素超标风险。实验数据表明,经Q膜处理后,最终蛋白浓缩原液的内毒素水平可稳定控制在0.5 EU/mL的严格标准以下(见图5 Case 2和Case 3),为生物制药企业提供了可靠的工艺保障。
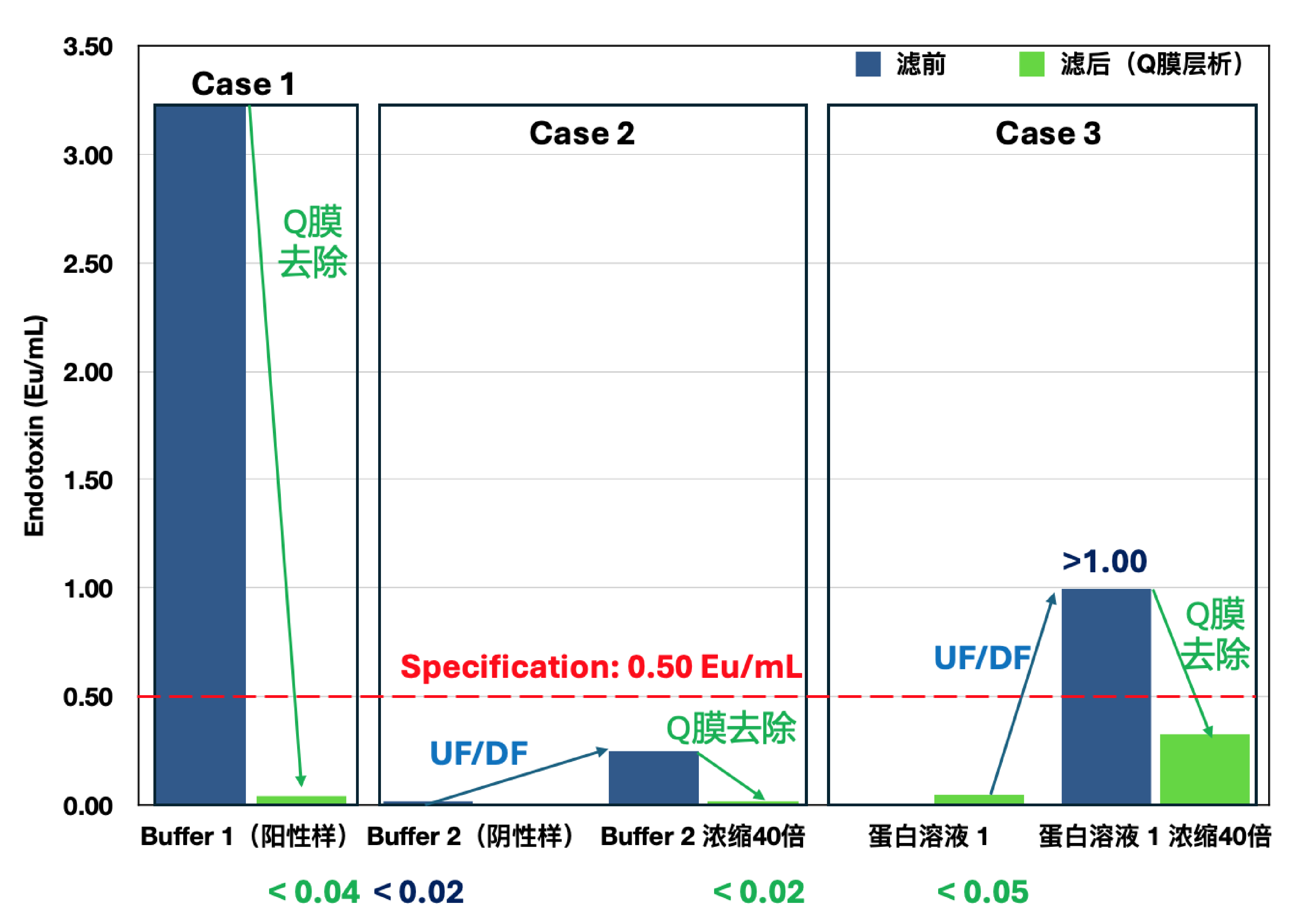
图5: Purcise™ Q强离子交换吸附膜在生物制药浓缩溶液内毒素去除方面的表现。Case 1)Buffer1(阳性样)为UF/DF Buffer(Tris 和甘露糖成份)加入标准内毒素原液至终浓度为3 EU/ml; 工艺参数:90mL Buffer 1经过0.9 mL Purcise™ Q针头式膜吸附器以9 mL/min流速进行过滤. 滤后内毒残留:<0.04 EU/ml。Case 2)Buffer 2为UF/DF换液Buffer(Tris 、甘露糖和吐温80成份)工艺参数:2 L Buffer经过5 mL Purcise™ Q膜吸附器膜以10 MV/min过滤,得到滤后液,对滤后液进行超滤浓缩。Buffer滤后浓缩液内毒残留:<0.02 EU/ml。Case 3)Q膜去除步骤:A). 使用0.9 ml Purcise™ Q针头式膜吸附器按流速4.5 ml/min对1L蛋白溶液1样品(浓度:~ 8.0 g/L)进行过滤,去除到蛋白样品中游离内毒素,滤后内毒残留:<0.05 EU/ml。B). UF/DF buffer准备:使用5 ml Purcise™ Q膜吸附器按流速50 ml/
挑战二:PUPSIT无菌保障与颗粒控制
随着全球药品监管标准的持续升级,PIC/S GMP Annex I明确将PUPSIT(Pre-Use Post Sterilisation Integrity Testing,使用前灭菌后完整性测试)列为强制性要求。该标准覆盖从灌装到包装的全流程无菌保障体系,使得PUPSIT验证与颗粒物管控成为GMP审计的核心关注点。值得注意的是,2025年3月中国国家药监局发布的《药品生产质量管理规范(2010年修订)》无菌药品附录(征求意见稿)已与国际标准接轨,首次将PUPSIT纳入法规要求体系:
“第一百八十七条 除菌过滤器组件应当在使用前灭菌后进行完整性测试。用于药液除菌的除菌过滤器,应当在使用后从外壳中取出前进行完整性测试。应当采用经验证的方法进行完整性测试,依据过滤系统验证确定的标准对测试结果进行判定。”
• 一次性使用PUPSIT系统
过去几年,制药行业对PUPSIT(使用前灭菌后完整性测试)的适用性一直存在技术争议,特别是在一次性使用系统领域。争议的核心焦点在于"使用后料液中的杂质是否可能掩盖滤膜缺陷"。2020年,PDA(注射剂协会)在其权威期刊发表的研究论文证实,这种潜在的掩盖效应确实与料液的成分复杂程度呈正相关。虽然生物制药生产过程中的料液通常具有较高的纯净度,理论上不应存在明显的缺陷掩盖风险,但欧盟监管机构仍坚持认为PUPSIT是确保无菌保证的关键控制措施。这一立场反映了监管机构对风险控制的谨慎态度,即使是在低风险场景下也要求采取最高标准的保障措施。
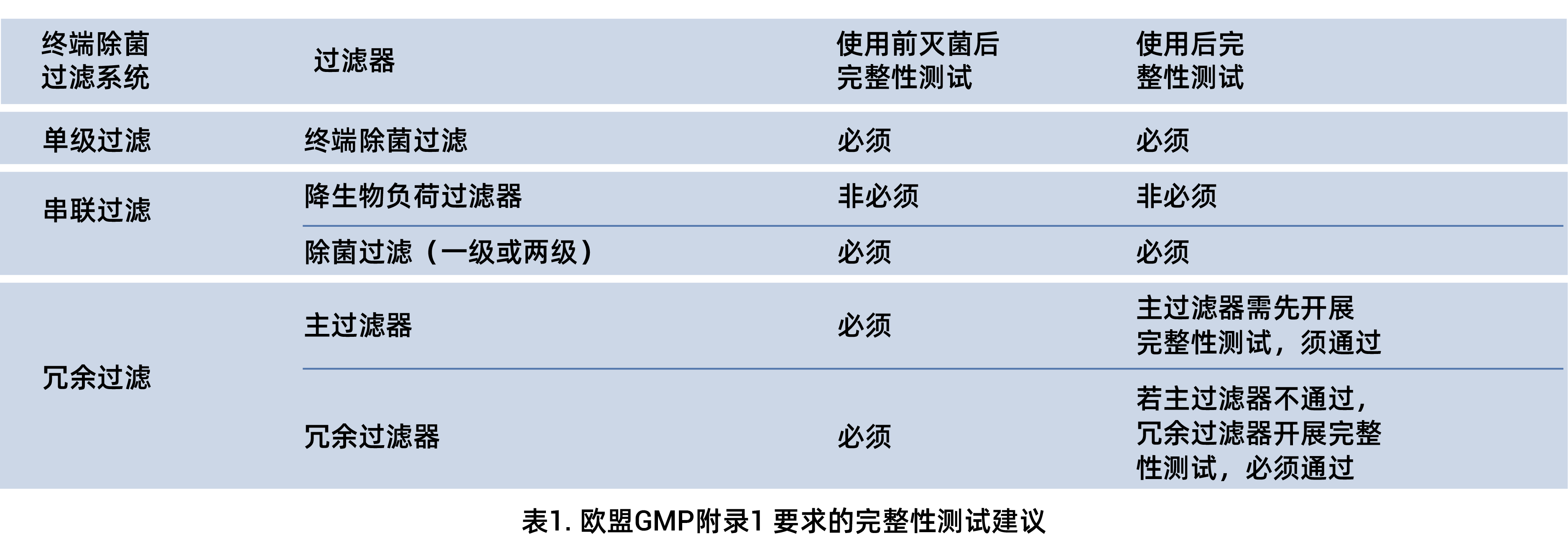
Cobetter Lifecube SAP 一次性使用 PUPSIT系统可有效解决使用前灭菌后完整性测试的难题。根据欧盟无菌附录Annex I,滤器是否需要开展完整性测试,可参考表1。

• 不锈钢系统的无菌保障:屏障过滤器
在不锈钢系统的PUPSIT测试实施过程中,业界还存在一个关键性技术争议:测试操作本身可能破坏已灭菌滤器的完整性,反而增加制剂的无菌保障风险。这一争议源于测试过程中必须进行的废液排放环节——废液管路通常需接入厂区排水系统,而排水管道系统可能存在严重的微生物污染风险。在排放操作时,排水系统的气溶胶和微生物可能通过废液管路发生逆向污染,对已灭菌的过滤系统造成二次污染。
针对这一行业痛点,科百特基于多年膜科学技术积累,成功攻克技术难关,研发推出Cobetter Ultcap™囊式过滤器解决方案。该产品创新性地集成了排水排气双重功能,同时具备反向无菌屏障特性,有效解决了传统排水系统的污染风险。这一突破为制药企业提供了符合国际标准且更具性价比的选择,完整供应链安全保障,助力企业更好地满足日益严格的GMP合规要求。
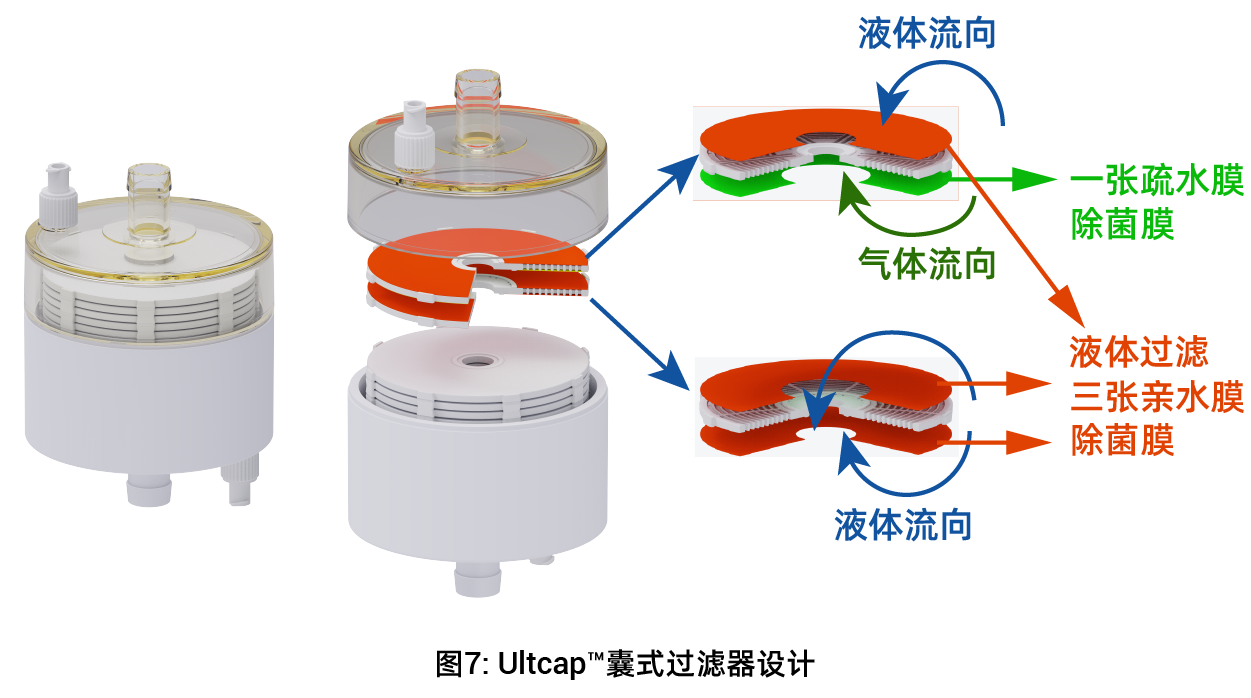
Ultcap™囊式屏障过滤器与筒式屏障过滤器用于优化无菌过滤系统在使用前完整性测试。产品使用PVDF膜具有高通量,高流速,低蛋白吸附,多类型试剂兼容性等特点。 产品因特殊的圆盘层叠过滤结构设计将亲水0.2 μm 除菌级PVDF膜与疏水0.2 μm 除菌级PVDF膜同时组合到单个过滤器,此结构可同时无菌过滤气体与液体。
应用方向
1:无菌系统中使用前灭菌后完整性测试 (PUPSIT)
2:灭菌后冷却系统排气使用
3:过滤器冲洗期间对无菌系统排气排液
• 一次性系统颗粒控制-灌装系统
一次性系统的颗粒控制一直困扰着生物制药行业,颗粒物控制不是简单的源头控制和trouble shooting,而是需要系统性工程化质量控制解决方案,这也是难点所在。科百特从膜材、设计、组装环境、清洗工艺、及过程检测全流程开展质量控制,成功将颗粒物水平降低1-2个数量级。


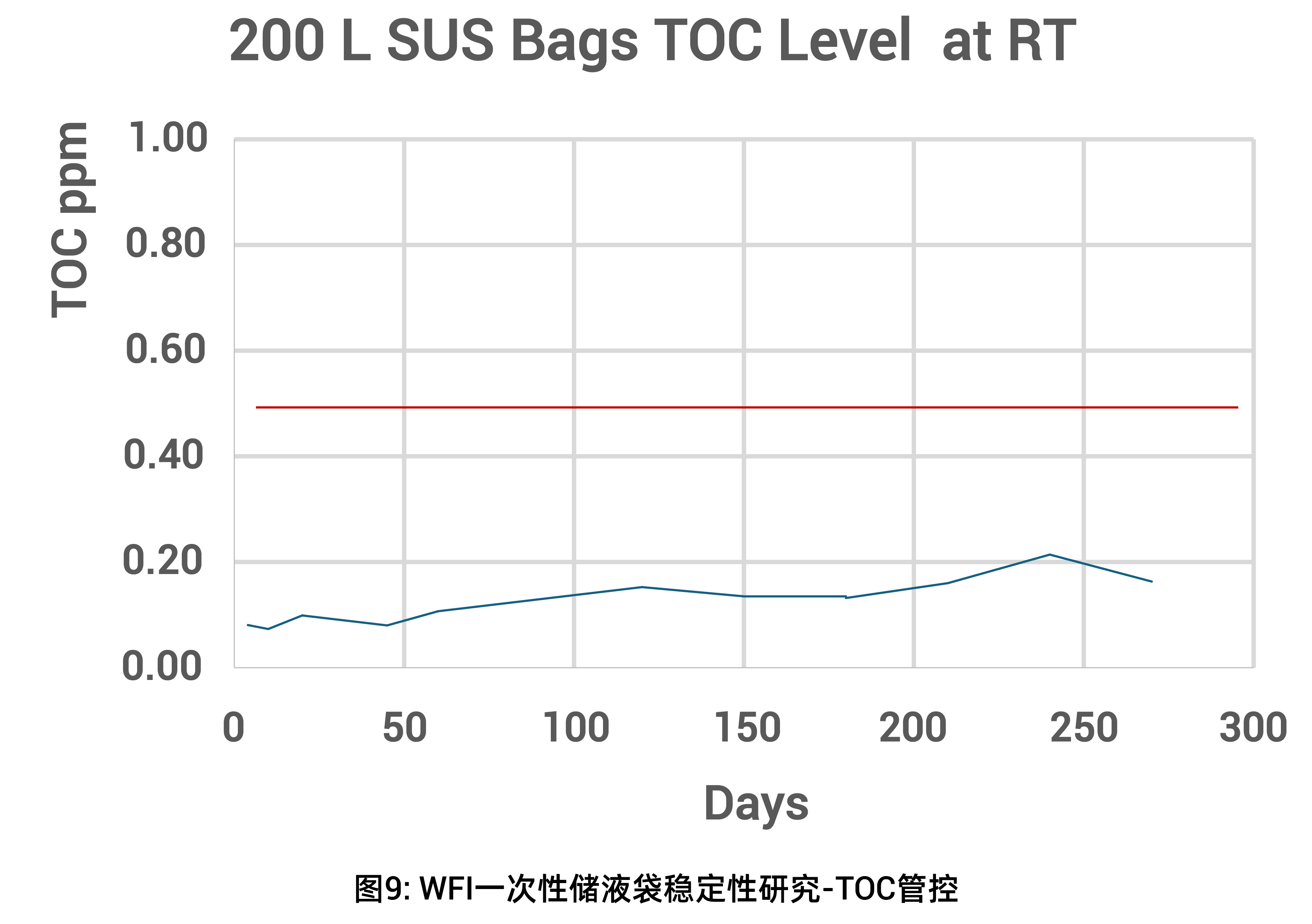
挑战三:注射用水储存过程中超低TOC控制要求
欧洲药典对注射用水(WFI)设定了严格的500ppb总有机碳(TOC)限值标准。虽然WFI原水可以达到这一要求,但在使用γ射线辐照灭菌的一次性储液袋系统时,WFI与PE膜、硅胶管、热塑管等组件接触后常出现TOC升高现象。这主要源于辐照过程中塑料材料会分解产生挥发性小分子有机物,溶入WFI后导致TOC超标。
开发超低TOC的一次性储液系统需要采用质量源于设计(QbD)理念,从原材料筛选、产品设计到生产工艺进行全方位优化,以满足5L-200L不同规格WFI储液需求。其中,小容量储液袋因接触比表面积大,技术难度更高。
Cobetter Lifecube 一次性储液袋系列经严格验证,可确保WFI储存期间TOC达标。针对5L等特殊规格,公司还开发了"半导体级洁净度"储液袋,为高端应用提供定制解决方案。